TP11: Impact of stress on aging of BDNF-dependent synaptic and cognitive functions (Volkmar Leßmann)
 During aging many biological and environmental processes contribute to the functional decline of cells. However, the mechanisms how this aging is reflected on the level of altered synaptic plasticity is unknown. Interestingly, there is a wide heterogeneity in the pace of aging in human and animal populations (Foebel, Pedersen, 2016). One factor that likely contributes to accelerated aging processes is the exposure to adverse environmental events in early and/or later life (Gassen et al., 2017). There is strong evidence that stress-induced alterations in BDNF expression contribute to long-lasting cellular and functional alterations in the hippocampal network (Bath et al., 2013). Stressful events alter spine densities in areas CA3 and CA1 of the hippocampus (Duman and Duman, 2015) and result in long-lasting reductions in BDNF protein expression (Lakshminarasimhan and Chattarji, 2012). Since BDNF is a key mediator of synaptic plasticity and exerts neuroprotective functions, reduced BDNF levels and subsequently altered activation of downstream signaling molecules (Panja et al., 2014) might be a key mechanism for accelerated aging of synaptic transmission. We recently observed a decline of brain BDNF levels and abundance of signaling-competent flTrkB receptors in aged WT mice (Petzold et al., 2015). In BDNF+/- mice this additional age-dependent decline leads to deficits in hippocampus- and amygdala-dependent learning processes (Psotta et al., 2013; Endes and Lessmann, 2012;).
During aging many biological and environmental processes contribute to the functional decline of cells. However, the mechanisms how this aging is reflected on the level of altered synaptic plasticity is unknown. Interestingly, there is a wide heterogeneity in the pace of aging in human and animal populations (Foebel, Pedersen, 2016). One factor that likely contributes to accelerated aging processes is the exposure to adverse environmental events in early and/or later life (Gassen et al., 2017). There is strong evidence that stress-induced alterations in BDNF expression contribute to long-lasting cellular and functional alterations in the hippocampal network (Bath et al., 2013). Stressful events alter spine densities in areas CA3 and CA1 of the hippocampus (Duman and Duman, 2015) and result in long-lasting reductions in BDNF protein expression (Lakshminarasimhan and Chattarji, 2012). Since BDNF is a key mediator of synaptic plasticity and exerts neuroprotective functions, reduced BDNF levels and subsequently altered activation of downstream signaling molecules (Panja et al., 2014) might be a key mechanism for accelerated aging of synaptic transmission. We recently observed a decline of brain BDNF levels and abundance of signaling-competent flTrkB receptors in aged WT mice (Petzold et al., 2015). In BDNF+/- mice this additional age-dependent decline leads to deficits in hippocampus- and amygdala-dependent learning processes (Psotta et al., 2013; Endes and Lessmann, 2012;).
However, the mechanisms how BDNF and its downstream signaling molecules are involved in these processes are widely unknown, thus limiting the exploitation of its possible therapeutic potential against such aging processes. Identifying altered signaling events and protein interaction partners downstream of TrkB activation are likely to provide novel approaches to reverse stress-induced changes that result in accelerated aging.
Hypothesis: We hypothesize that stressful periods occurring during different stages of life, e.g. in youth, adult-hood or both (as 2-hit model), result in accelerated aging of synaptic transmission and synaptic plasticity by altering BDNF/TrkB and proBDNF/p75NTR signaling, ultimately causing even enhanced decline in learning associated synaptic plasticity, memory performance, and cognition in aged animals that experienced stress earlier in their life. Aims:
- To analyze how stressful events experienced in early life and in adulthood, impact long-term synaptic plasticity at hippocampal synapses and how they affect hippocampus-dependent learning in vivo.
- To unravel stress- and aging-dependent alterations in interaction partners downstream of TrkB using biochemical and proteomic analyses.
- To identify treatments counteracting stress-induced accelerated aging processes, by either chronically or acutely increasing BDNF expression.
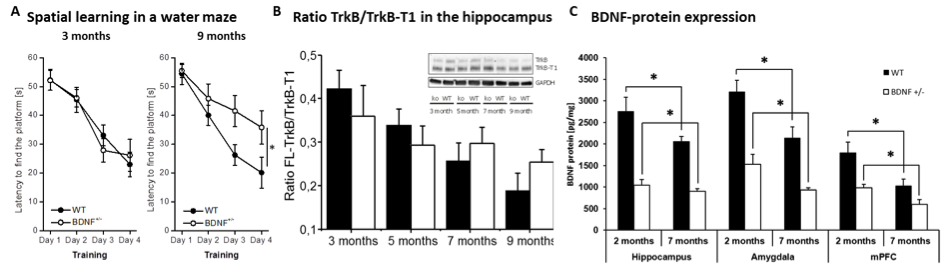
A: Heterozygous BDNF knockout animals exhibit an age-dependent deficit in spatial learning. B: The abundance of signaling-competent TrkB receptors declines with aging. C: BDNF protein expression declines in different areas with aging.
Collaborations: TP1 Dieterich (dendritic proteome of BDNF/TrkB downstream signaling/ interaction partners) TP10 Stork (stress related network activity and signaling in memory decline), TP3 Kreutz (molecular mechanisms of BDNF-dependent altered synaptic plasticity) TP6 Dityatev (electrophysiological analysis of LTP in hippocampal slices from aged mice).